
-
Introduction
Poly(ethylene terephthalate), more commonly known as PET in the packaging industry and generally referred to as ‘polyester‘ in the textile industry, is an indispensable material with immense applications owing to its excellent physical and chemical properties. On the other hand, due to its increasing consumption and non-biodegradability, PET waste disposal has created serious environmental and economic concerns. Thus, management of PET waste has become an important social issue. In view of the increasing environmental awareness in the society, recycling remains the most viable option for the treatment of waste PET. Among the various methods of PET recycling (primary or ‘in-plant’, secondary or mechanical, tertiary or chemical, quaternary involving energy recovery), only chemical recycling conforms to the principles of sustainable development because it leads to the formation of the raw materials from which PET is originally made. Chemical recycling utilizes processes such as hydrolysis, methanolysis, glycloysis, ammonolysis and aminolysis. In a large collection of researches for the chemical recycling of PET, the primary objective is to increase the monomer yield while reducing the reaction time and/or carrying out the reaction under mild conditions. Continuous efforts of researchers have brought great improvements in the chemical recycling processes. This paper reviews methods for the chemical recycling of PET with special emphasis on glycolytic depolymerization with ethylene glycol. It covers the researches, including the works by the authors, on various processes and introduces recent developments to increase monomer yield. Processes including sub- & supercritical, catalytic, and microwave-assisted depolymerization are discussed. This paper also presents the impact of the new technologies such as nanotechnology on the future developments in the chemical recycling of PET.
1.1 PET: Synthesis and properties
PET is a polycrystalline polyester formed from the esterification of terephthalic acid (TPA) with ethylene glycol (EG) or from the transesterification of dimethyl terephthalate (DMT) with EG. Synthesis of PET from either process involves two reaction steps as shown in Fig. 1. The first step (Figs 1a, 1b) is the formation of an intermediate monomer bis(2-hydroxyethyl terephthalate) (BHET) with the release of a small molecule, which is either water or methanol. The second is the polycondensation of BHET to produce PET in melt phase with the release of EG under high vacuum (Scheirs, 1998; Scheirs & Long, 2003).
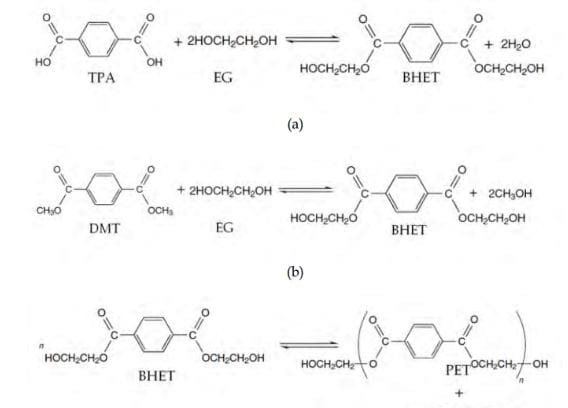
Fig. 1. Reaction scheme for PET synthesis. BHET is first formed from the reaction of either (a) TPA and EG, or (b) DMT and EG, and (c) eventually polymerized to PET.
As a thermoplastic polyester resin, PET exhibits interesting physical and chemical properties. It is an amorphous glass-like material in its purest form. Crystallinity in PET can be enhanced by adding modifying additives or by heat treatment of the polymer melt. PET is classified as a semi-crystalline polymer, and when heated above 72 oC, it changes from a rigid glass-like state into a rubbery elastic form where the polymer molecular chains can be stretched and aligned in either one direction to form fibers, or in two directions to form films and bottles. If PET is held in the stretched form at temperatures above 72 ⁰C, it slowly crystallizes and the material starts to become opaque and less flexible. It is then known as crystalline PET. Meanwhile, if the melt is cooled quickly while still in stretched state, the chains are frozen with their original orientation. The resulting material is an extremely tough plastic, typical of a PET bottle (Sinha et al., 2008). Commercial PET melts between 255 Recent Developments in the Chemical Recycling of PET 67 and 265 ⁰C, while more crystalline PET melts at 265 ⁰C. Virgin PET is capable of morphological and structural reorganization, which is attributed to its multiple endothermic transitions. This leads to better crystal structures as temperature increases (Awaja & Pavel, 2005).
1.2 Applications, production and issues
Because of its low cost (Thompson et al., 2009), excellent tensile strength, chemical resistance, clarity, processability, and reasonable thermal stability (Caldicott, 1999), PET has been used in a wide range of applications. The demand and usage of PET worldwide according to application is summarized in Table 1 (Scheirs & Kaminsky, 2006). It is mainly applied in the textile industry, where more than 60% of all the PET produced worldwide is consumed. Enormous amounts are also used for other applications including manufacture of video and audio tapes, X-ray films, thermoformed products (e.g. material handling equipments, house-wares, automobile products, lighting products, sporting goods, etc) and food packaging (Carraher, 2000; ILSI Europe, 2000; Olabisi, 1997). In food packaging, PET has become the choice especially for beverages mainly due to its glass-like transparency coupled with adequate gas barrier properties for retention of carbonation. It provides an excellent barrier against oxygen and carbon dioxide in the carbonated soft drink sector, which has been growing more rapidly than other applications. In addition, it exhibits a high toughness/weight property ratio, which allows lightweight and securely unbreakable containers with large capacity (Welle, 2011).

Table 1. The global demand and future prediction of PET by application. ( Unit in thousand tons).
From its main applications, PET is mainly classified as fiber-grade or bottle-grade. These grades differ mainly in molecular weights, intrinsic viscosity, optical appearance, and production recipes. Fiber-grade PET has a number-average molecular weight (MWn) of 15,000 to 20,000 g/mol and intrinsic viscosity (IV) of 0.40 to 0.70 dL/g. Bottle-grade PET average molecular weight ranges from 24,000 to 36,000 g/mol and IV from 0.70 to 0.85 dL/g. (Awaja & Pavel, 2005; Gupta & Bashir, 2002).
PET‘s popularity has risen tremendously since it discovery in the early 1940s. In the year 2000, the global PET production capacity exceeded 33 million metric tons per year (Rieckmann, 2003). The total global consumption has risen from 11.8 million metric tons in 1997 (Paszun & Spychaj, 1997) to 23.6 million in 2005 (Pohler, 2005, as cited in Karayannidis & Achilias, 2007) and 54 million in 2010 (IHS, 2011). It is expected to grow by 4.5% per year from 2010 to 2015. In Europe and America, the rise of PET consumption is mainly 68 Material Recycling – Trends and Perspectives maintained by PET bottle production while in Asia, the expansion of PET use is related to the higher production of fibers, due to the shift of fiber production from the industrialized countries to low-wage countries.
Along with the widespread application of PET is the inevitable creation of large amounts of PET waste. PET does not have any side effects on the human body, and does not create a direct hazard to the environment. However, due to its substantial fraction by volume in the waste stream and its high resistance to the atmospheric and biological agents, it is considered as a noxious material (Paszun & Spychaj, 1997). With the increase in the amount of PET wastes, its disposal began to pose serious economical and environmental problems.
In view of the increasing environmental awareness in the society, recycling remains the most viable option for the treatment of waste PET. Environmental and economic considerations as well as energy conservation issues pushed the wide-scale recycling of PET (Nir et al., 1993); it was not simply a trend or a new marketing strategy to make a profit (Grasso, 1995, as cited in Shukla & Kulkarni, 2002). The recycling of PET does not only serve as a partial solution to the solid waste problem but also contributes to the conservation of raw petrochemical products and energy. Products made from recycled plastics can result in 50-60% capital saving as compared to making the same product from virgin resin (Sinha et al., 2008).
Nevertheless, Welle noted that the main driving force in PET recycling is not cost reduction, but the business sector’s embracing of sustainability ethics and the public’s concern about the environment (Welle, 2011).
- PET recycling methods
PET is considered one of the easiest materials to recycle, and is second only to aluminum in terms of the scrap values for recycled materials (Shceirs, 1998). Because of this, PET recycling has been one of the most successful and widespread among polymer recycling (Karayaniddis et al., 2006; Karayaniddis & Achilias, 2007). PET recycling methods can be categorized into four groups namely primary, secondary, tertiary, and quaternary recycling There is also a so called ‘zero-order‘ recycling technique, which involves the direct reuse of a PET waste material (Nikles & Farahat, 2005). There are many other terminologies used for these plastic recycling categories; Hopewell and his colleagues have summarized these different terminologies (Hopewell et al., 2009).
2.1 Primary recycling
Primary recycling, also known as re-extrusion, is the oldest way of recycling PET. It refers to the ‘‘in-plant’’ recycling of the scrap materials that have similar features to the original products. This process ensures simplicity and low cost, but requires uncontaminated scrap, and only deals with single-type waste, making it an unpopular choice for recyclers (AlSalem, 2009; Al-Salem et al., 2009).
2.2 Secondary recycling
Secondary recycling, also known as mechanical recycling, was commercialized in the 1970s.
It involves separation of the polymer from its contaminants and reprocessing it to granules via mechanical means. Mechanical recycling steps include sorting and separation of wastes, Recent Developments in the Chemical Recycling of PET 69 removal of contaminants, reduction of size by crushing and grinding, extrusion by heat, and reforming (Aguado & Serrano, 1999). The more complex and contaminated the waste is, the more difficult it is to recycle mechanically. Among the main issues of secondary recycling are the heterogeneity of the solid waste, and the degradation of the product properties each time it is recycled. Since the reactions in polymerization are all reversible in theory, the employment of heat results to photo-oxidation and mechanical stresses, causing deterioration of the product’s properties. Another problem is the undesirable gray colour resulting from the wastes that have the same type of resin, but of different color.
2.3 Tertiary recycling
Tertiary recycling, more commonly known as chemical recycling, involves the transformation of the PET polymer chain. Usually by means of solvolytic chain cleavage, this process can either be a total depolymerization back to its monomers or a partial depolymerization to its oligomers and other industrial chemicals. Since PET is a polyester with functional ester groups, it can be cleaved by some reagents such as water, alcohols, acids, glycols, and amines. Also, PET is formed through a reversible polycondensation reaction, so it can be transformed back to its monomer or oligomer units by pushing the reaction to the opposite direction through the addition of a condensation product. These low molecular products can then be purified and reused as raw materials to produce highquality chemical products (Carta et al., 2003).
Among the recycling methods, chemical recycling is the most established and the only one acceptable according to the principles of ‘sustainable development‘, defined as development that meets the needs of present generation without compromising the ability of future generations to meet their needs (Harris, 2001; World Commission on Environment and Development, 1987), because it leads to the formation of the raw materials (monomers) from which the polymer is originally made. In this way the environment is not surcharged and there is no need for extra resources for the production of PET (Achilias & Karayannidis, 2004).
The reaction mechanism for PET depolymerization consists of three reversible reactions. First, the carbonyl carbon in the polymer chain undergoes rapid protonation where the carbonyl oxygen is converted to a second hydroxyl group. Second, the hydroxyl oxygen of the added hydroxyl-bearing molecule slowly attacks the protonated carboxyl carbon atom.
Third, the carbonyl oxygen (which was converted to hydroxyl group in the first step) and a proton are rapidly removed to form water or a simple alcohol and the catalytic proton (Patterson, 2007).
As shown in Fig. 2 (Janssen & van Santen, 1999), there are three main methods in PET chemical recycling depending on the added hydroxyl bearing molecule: glycol for gylcolysis, methanol for methanolysis, and water for hydrolysis. Other methods include aminolysis and ammonolysis. It has been five decades since the start of PET chemical recycling research, when patents were filed by Vereinigte Glanzstoff-Fabriken in the 1950s (Vereinigte Glanzstoff-Fabriken, 1956, 1957). Since then, numerous researches have been done in order to fully understand the chemical pathways of the depolymerization methods, and improve desired products yield from these methods.
70 Material Recycling – Trends and Perspectives
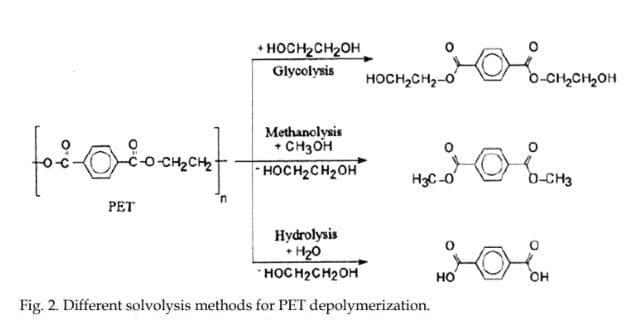
2.3.1 Hydrolysis
Hydrolysis involves the depolymerization of PET to terephthalic acid (TPA) and ethylene glycol by the addition of water in acidic, alkaline or neutral environment. The hydrolysis products may be used to produce virgin PET, or may be converted to more expensive chemicals like oxalic acid (Yoshioka et al., 2003). Concentrated sulfuric acid is usually used for acid hydrolysis (Brown & O’Brien, 1976; Pusztaszeri, 1982; Sharma et al., 1985; Yoshioko et al., 1994, 2001), caustic soda for alkaline hydrolysis (Alter, 1986), and water or steam for neutral hydrolysis (Campanelli et al., 1993,1994a; Mandoki, 1986). Hydrolysis is slow compared to methanolysis and glycolysis, because among the three epolymerizing agents (i.e. water, methanol, ethylene glycol), water is the weakest nucleophile. It also uses high temperatures and pressures. Another disadvantage of hydrolysis is the difficulty of recovery of the TPA monomer, which requires numerous steps in order to reach the required purity.
2.3.2 Methanolysis
Methanolysis is the degradation of PET to dimethyl terephthalate (DMT) and EG by methanol. Disadvantages of this method include the high cost associated with the separation and refining of the mixture of the reaction products. Also, if water perturbs the process, it poisons the catalyst and forms various azeotropes. Before, methanolysis and glycolysis were the methods applied on a commercial scale (Paszun, 1997), but today, it is not used for PET production anymore, and the lack of usefulness of recovering DMT rendered the methanolysis of PET to become obsolete (Patterson, 2007).
2.3.3 Glycolysis
As shown in Fig. 3, glycolysis is carried out using ethylene glycol to produce bis(2-hydroxyethyl) terephthalate and other PET glycolyzates, which can be used to manufacture unsaturated resins, polyurethane foams, copolyesters, acrylic coatings and hydrophobic dystuffs. The BHET produced through glycolysis can be added with fresh BHET and the mixture can be used in any of the two PET production (DMT-based or TPA-based) lines.
Recent Developments in the Chemical Recycling of PET 71 Diethylene glycol (Karayannidis et al., 2006), triethylene glycol (Öztürk & Güçlü, 2005), propylene glycol (Güclü et al., 1998; Vaidya & Nadkarni, 1987), or dipropylene glycol (Johnson & Teeters, 1991, as cited in Sinha et al., 2008) may also be used as solvent in PET glycolysis.
Besides its flexibilty, glyclolysis is the simplest, oldest, and least capital-intensive process.
Because of these reasons, much attention has been devoted to the glycolysis of PET.
Numerous works have been published about PET glycolysis, wherein the reaction has been conducted in a wide range of temperature and time. The works involving this process, from 1960, when Challa started to investigate the polycondensation equilibrium of melt glycolysis (Challa, 1960; As cited in Patterson, 2007), up until now when researchers are focused on developing more efficient glycolysis catalysts and investigating on the applications of the glycolysis products, will be discussed in detail in the later part of this work.
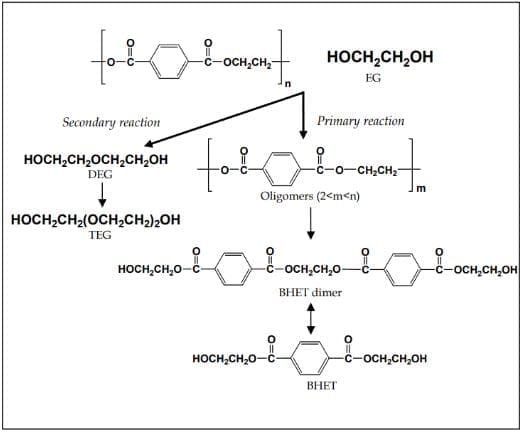
2.4 Quaternary recycling
Quaternary recycling represents the recovery of energy content from the plastic waste by incineration. When the collection, sorting and separation of plastics waste are difficult or economically not viable, or the waste is toxic and hazardous to handle, the best waste management option is incineration to recover the chemical energy stored in plastics waste in the form of thermal energy. However, it is thought to be ecologically unacceptable due to potential health risks from the air born toxic substances.
- Glycolytic depolymerization of PET
Studies on the kinetics of PET glycolysis (Campanelli et al., 1994b; J. Chen & L. Chen, 1999) have shown that glycolysis without a catalyst is very slow and complete depolymerization of PET to BHET cannot be achieved. It also yields an end product that contains significant amount of other oligomers in addition to the BHET monomer. This results in difficulty in recovering the BHET monomer when it is the desired product. Thus, research efforts have been directed towards increasing the rate and BHET monomer yield by developing highly efficient catalysts and other techniques, and optimizing the reaction conditions (e.g. temperature, time, PET/EG ratio, PET/catalyst ratio). Others sought for applications of the glycolysis product without the separation of oligomers (Grzebienek & Wesolowski, 2004).
Still others sought for more eco-friendly glycolytic process. Two decades after the beginning of PET glycolysis research, these efforts resulted in the significant increase in BHET monomer yield from just 65% with 8 hours reaction time to at least 90% with a significantly reduced reaction time of around 30 minutes.
3.1 Catalyzed glycolysis
The most studied method of increasing the glycolysis rate is catalysis. PET glycolysis is considered a transesterification reaction. Thus, transesterification catalysts have been applied to increase the reaction rate of PET glyclosis, with metal based catalysts being the most popular. Helwani et al. and Schuchardt et al. have listed all the catalysts that have been used before in other transesterification reactions (Helwani et al., 2009; Schuchardt et al., 1997).
Fig. 4 shows the reaction mechanisms of uncatalyzed glycolysis and that of glycolysis with metal-based catalyst (Shukla & Harad, 2005; Pingale et al., 2010). A free electron pair on the EG oxygen initiates the reaction by attacking the carbonyl carbon of the ester group of the polyester. The hydroxyethyl group of ethylene glycol then forms a bond with the carbonyl carbon of the polyester breaking the long chain into short chain oligomers and finally BHET.
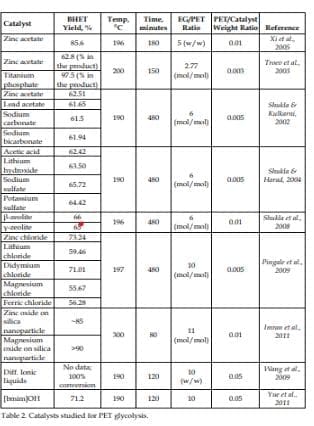
The rate of glycolysis reaction depends on a number of parameters including temperature, pressure, PET/EG ratio, and the type and amount of catalyst. Also, the transformation of dimer to BHET monomer is a reversible process. Prolonging the reaction after the equilibrium of the two is attained will cause the reaction to shift backwards, increasing the amount of dimer at the expense of the BHET monomer. It is thus important to know the optimum conditions of the glycolysis reaction. With metal based catalysts (Fig. 4b), the metal forms a complex with the carbonyl group, facilitating the attack of EG on PET leading to the formation of BHET. A number of glycolytic depolymerization processes have been reported with different catalysts and different reaction conditions. We have listed the catalysts studied to date in Table 2.
Recent Developments in the Chemical Recycling of PET 73.
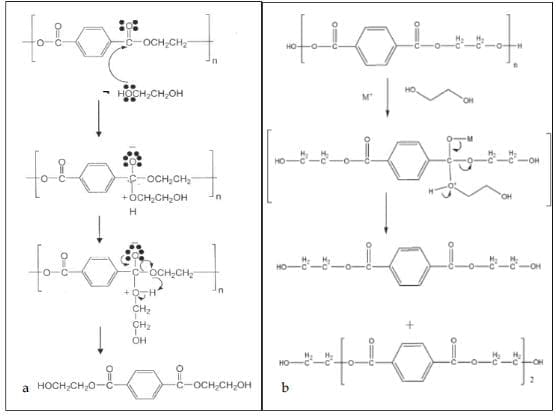
3.1.1 Metal salts
The oldest reported catalysts for PET glycolysis are metal acetates. Zinc acetate was first used by Vaidya and Nadkarni for their work dealing with the synthesis of polyester polyols from PET waste (Vaidya & Nadkarni, 1988). In 1989, Baliga and Wong further investigated the use of metal acetates (zinc, manganese, cobalt, and lead) as catalysts. They reported that zinc acetate showed best results in terms of the extent of depolymerization reactions of PET.
They also observed that the equilibrium between the BHET monomer and dimer was reached after 8 hours of reaction with the temperature at 190 ⁰C. This may be considered as the beginning of PET glycolysis catalysts research as several researches followed later.
Ghaemy and Mossaddegh verified the results obtained by Baliga and Wong, and the order of activity of the catalysts (Zn+2 > Mn+2 > Co+2 > Pb+2) (Ghaemy & Mossaddegh, 2005). J. Chen and L. Chen studied the kinetics of PET glycolysis with zinc acetate catalyst at the same temperature, and they found out that the equilibrium between the BHET monomer and the dimer was reached after two hours, as opposed to 8 hours from Baliga and Wong (J. Chen & L. Chen, 1999). Meanwhile, C. Chen studied that of manganese acetate and found out that the best glycolysis condition for the same temperature was the reaction time of 1.5 h with 0.025 mol/kg PET (C. Chen, 2003). Xi et al. investigated the optimum condition of the reaction at 196 ⁰C. They reported that a 3-hour reaction with EG/PET weight ratio of 5, and catalyst/PET weight ratio of 0.01 can deliver 85.6% BHET yield (Xi etal., 2005). Goje and Mishra also studied the optimum conditions of PET glycolytic depolymerization at 197 °C, and they reported 98.66% PET conversion with the reaction time of 90 minutes and PET particle size of 127.5 μm. The optimal PET particle size was the size at which PET weight loss was maximum. They did not measure the BHET yield though, because the reaction pathway they used produced DMT and EG instead of BHET (Goje & Mishra, 2003).
Dayang et al. later used the products from PET glycolysis catalyzed by zinc acete to make themally stable polyester resin via polyesterification with maleic anhydride and crosslinking with styrene (Dayang et al., 2006). The synthesis of unsaturated polyester resin actually dates back to 1964 (Ostrysz et al., 1964, as cited in Paszun & Spychaj, 1997). This unsaturated polyester resin was later reinforced with natural fibers in the study made by Tan et al. to produce a fiber composite with good mechanical properties (Tan et al., 2011).
Although metal salts are effective in increasing the PET glycolysis rate, it should be noted that zinc salts, and presumably other metal salts, have a catalytic effect on glycolysis of PET only below 245 °C, and apparently do not promote any further increase in the reaction rate above that temperature due to mass transfer limitations (Campanelli et al., 1994b). Thus, a need to develop new catalysts that can overcome this limitation. In 2003, Troev et al. introduced titanium (IV) phosphate as a new catalyst. They reported that glycolysis in the presence of the new catalyst was faster compared to that with zinc acetate. Their data showed that at 200°C, 150 minutes reaction time and 0.003 catalyst/PET weight ratio, the glycolyzed products from titanium (IV) phosphate catalyzed reaction consisted of 97.5% BHET, which was significantly higher than that of zinc acetate, which was 62.8 % (Troev et al., 2003).
Since lead and zinc are heavy metals known to have negative effects on the environment, Shukla’s group started to develop milder catalysts that are comparatively less harmful to the environment. They started with mild alkalies, sodium carbonate and sodium bicarbonate, and reported that the monomer yields (Refer to Table 2) were comparable with those of the conventional zinc and lead acetate catalysts (Shukla & Kulkarni, 2002). They also reported glacial acetic acid, lithium hydroxide, sodium sulfate, and potassium sulfate to have comparable yields (Table 2) with those of the conventional heavy metal catalysts (Shukla & Harad, 2005). They recently used the recovered BHET monomer to produce useful products such as softeners and hydrophobic dyes for the textile industry (Shuka et al., 2008, 2009).
López-Fonseca et al. also used these eco-friendly catalysts in their study of catalyzed glycolysis kinetics (López-Fonseca et al., 2010, 2011). The latest catalysts that Shukla’s group developed are inexpensive and readily available metal chlorides, wherein zinc chloride reportedly gave the highest BHET yield equal to 73.24% (Pingale et al., 2010).
3.1.2 High surface area catalysts: Nanocomposites
In 2008, Shukla et al. reported new addition to their set of eco-friendly catalysts in the form of zeolites (Shukla et al., 2008). Zeolites have been used as catalysts in other reactions before, and their catalytic activity can be credited to their large surface area in mesopores and micropores that provide numerous active sites. Their result, however, showed that the BHET yield (Table 2) did not deliver any significant improvement from the other catalysts they previously reported.
Looking back to the number of catalysts previously discussed in this work, it is noticeable that the BHET yield never reached the 90% mark. The restricted amount of BHET yield may be because the reaction was not performed at temperatures above 245⁰C, since the previously reported catalysts lose their effectiveness at increased temperatures anyway.
With the aim of increasing the BHET monomer yield at reduced reaction time, our group developed catalysts that are highly selective and can work at elevated temperatures – metal oxide catalysts. Metal oxides as glycolysis catalysts could provide a better alternative to conventional catalysts in that they have high mechanical strength, are thermally stable, and are cost effective. Metal oxides were used for other transesterification reactions before (Helwani et al., 2009; Singh & Fernando, 2007), but they had not been applied in PET glycolysis. In order to increase the metal oxide catalysts’ efficiency, we tried to increase the surface area of active sites by fabricating them at nanoscale. Besides increasing the surface area of the active sites, it is known that at nanoscale, the intrinsic properties of the catalysts may change, leading to increased effectiveness compared to that of their bulk conterpart (Heiz & Landman, 2007; Niederberger & Pinna, 2009). Fig. 5 (Imran et al., 2011; Wi et al, 2011) shows TEM images of the fabricated 60 nm (a) and 150 nm (b) silica nanoparticle used as supports and the supports with the deposited metal oxide catalysts. The metal oxide catalysts were deposited on the silica nanoparticle supports via a simple ltrasound assisted precipitation method. Good deposition was observed especially for cerium oxide and manganese oxide.
The oxides of zinc, manganese, and cerium deposited on silica anoparticle support were used as catalysts in a glycolytic reaction performed at 300 °C and 1.1 MPa with EG/PET molar ratio of 11, and PET/catalyst weight ratio of 0.01. The reaction reached equilibrium after 80 minutes, and the highest BHET yield reached more than 90%. Moreover, we found out that the smaller the size of the support is, the better is the distribution of the catalysts on the support. This could be due to the higher chances of contact between the catalyst and the support because of the higher surface-area-to-volume ratio for smaller supports. The better distribution of the catalysts resulted in higher catalytic activity.
3.1.3 Recyclable catalyst: Ionic liquids
It has not been long since ionic liquids were applied as catalyst for PET glycolysis when Wang et al. initiated the study and first reported its use in 2009 (Wang et al., 2009a). The main advantage of ionic liquids over conventional catalysts like metal acetates is that the purification of the glycolysis products is simpler.
They prepared different ionic liquids and performed glycolysis reactions in the presence of these ionic liquids at atmospheric pressure with different temperature and time. 100 % conversion of PET was achieved after 8 hours at a temperature of 180 °C, with the 1-butyl-3-methylimidazolium bromide ([bmim] Br) being the best catalyst in terms of PET conversion & ease and cost of preparation. They concluded that the BHET purity from their method was high. They did not, however quantitatively measure the BHET yield from their experiment.
After this, they extended their research by investigating the reusability of the ionic liquid catalysts and kinetics of the PET degradation by ionic liquid alone. They concluded that the catalysts can be used repeatedly, that the degradation reaction is first-order with activation energy equal to 232.79 kJ/mol, and that it can potentially replace the traditional organic solvents used in PET degradation (Wang et al., 2009b). Recently, they successfully applied Fecontaining magnetic ionic liquid as a catalyst for PET glycolysis. They reported that this catalyst has better catalytic activity than the conventional metal salts or the pure ionic liquid with the amount of catalyst affecting the PET conversion and BHET selectivity (Wang et al., 2010). Yue et al. followed this study by using basic ionic liquid, and reported that basic [bmim]OH exhibits higher catalytic activity than [bmim] Br and [bmim] Cl.
Fig. 5. TEM images of the (a) 60 nm silica support fabricated via water-in-oil microemulsion method, (b) 150 nm silica support from Stober method, (c) ZnO on 60 nm silica support, (d) ZnO on 150 nm silica support, (e) CeO2 on 60 nm silica support, and (f) Mn3O4 on 150 nm silica support.
They attained 100 % PET conversion with 71.2% BHET yield by performing the glycolysis at 190 ⁰C for 2 hours with EG/PET molar ratio of 10 and catalyst/PET weight ratio of 0.05 (Yue et al., 2011). As can be deduced in this study, the recoverability and reusability of ionic liquid catalyst permits the use of higher amount of catalyst.
3.2 Solvent-assisted glycolysis
In 1997, Güçlü et al. added xylene in the zinc acetate catalyzed PET glycolysis reaction, and obtained 80% BHET yield, which was higher than the yield from that without xylene. The main objective of xylene was initially to provide mixability to the PET-glycol mixture. At temperatures between 170 ⁰C and 225 ⁰C, EG dissolves sparingly in xylene while it dissolves readily in PET. Meanwhile, the glycolysis products are soluble in xylene. Therefore, as the reaction progressed, the glycolysis products moved from the PET-glycol phase to the xylene phase, shifting the reaction to the direction of depolymerization (Güçlü et al., 1997). Sole publication is available for this PET glycolysis technique. Further investigations may have been prevented by the reason that organic solvents are harmful to the environment and massive use of these solvents is not a very attractive idea.
3.3 Supercritical Glycolysis
The use of supercritical conditions has been explored earlier in PET hydrolysis (Sato et al., 2006) and methanolysis (Minoru et al., 2005; Yang et al., 2002), but only recently for glycolysis (Imran, et al., 2010). The main advantage of the use of supercritical fluids in a reaction is the elimination of the need of catalysts, which are difficult to separate from the reaction products. It is also environment friendly. Our group investigated the use of EG in its supercritical state (Tc = 446.70 ⁰C, Pc = 7.7 MPa) (Imran et al., 2010). Supercritical process was carried out at 450 ⁰C and 15.3 MPa, and the results were compared with those from the
subcritical processes carried out at 350 ⁰C and 2.49 MPa, and 300 ⁰C and 1.1 MPa. Compared to the subcritical process, the BHET-dimer equilibrium was achieved much earlier for supercritical process: a maximum BHET yield of 93.5 % was reached in mere 30 minutes.
Owing to high temperature and pressure, supercritical glycolysis delivered a very high yield of BHET while suppressing the yield of the side products (0.69% DEG yield and almost negligible formation of oligomers, BHET dimer, and TEG). If economically feasible, supercritical glycolysis may be able to replace catalyzed glycolysis.
3.4 Microwave-assisted glycolysis
Beyond eco-friendly catalysts, Pingale and Shukla extended their study to the use of unconventional heating source of microwave radiations. The employment of microwave radiations as heating source drastically decreased the time for the completion of reaction from 8 hours to just 35 minutes. However, it did not increase the BHET monomer yield (Pingale and Shukla, 2008). The use of more efficient catalyst along with microwave irradiation heating may be able to increase the BHET yield while decreasing the reaction time.
-
Conclusion: Challenges and opportunities
From the discovery of PET in 1940s and the start of PET chemical recycling in 1950s that attracted great interest from the research community, PET glycolysis has gone a long way, Recent Developments in the Chemical Recycling of PET 79 back when zinc acetate was first used as catalyst to obtain about 60% BHET yield after 8 hours of reaction until when silica nanoparticle-supported metal oxide catalysts were applied to obtain at least 90% yield after 80 minutes. Studies have already dealt with most of the problems dealing with PET glycolysis, including unfeasibility of operation due to long reaction times, low yields, severe conditions, and pollution problems. Researchers have developed catalysts to increase the rate and BHET monomer yield, catalysts that are environmentally friendly, catalysts that can be recovered and reused, a method that does not require catalysts, and many others.
However, PET glycolysis is still far from its peak. Though researchers have found ways to solve each problem separately, there is still no way to solve them all simultaneously. For instance, eco-friendly catalysts deliver lower yields compared to the not-so-eco-friendly ones (e.g. metal oxides). The main challenge that stands now is to deliver an efficient, sustainable, environment friendly, and less energy demanding way to chemically recycle PET. This may be an opportunity for researchers try to develop efficient and highly selective catalysts that can be recovered and reused. There may be many other ways to break the boundaries, and with the rapid advancement of technologies like nanotechnology, solutions may be discovered in the near future. We believe that by exploring the possibilities of technologies that have not yet been applied, great advancements on PET glycolysis can be made. For instance, it has been reported that ultrasound can induce the scission of polymer chains (Kuijpers et al., 2004). Ultrasound assisted depolymerization has been applied to other depolymerization processes before (Sayata & Isayev, 2002; Sayata et al., 2004; Shimetal., 2002), but it has not been explored in PET glycolysis yet. Nanotechnology, which is growing by leaps and bounds may also be exploited to develop more highly efficient glycolytic depolymerization of PET.
-
Acknowledgement
This work was supported by the Resource Recyling R&D Center sponsored by 21C Frontier R&D Program, the Center for Ultramicrochemical Process Systems sponsored by KOSEF, the Basic Science Research Program through a National Research Foundation of Korea (NRF) grant funded by the Ministry of Education, Science and Technology (2010-0025671).
-
References
Achilias, D. & Karayannidis, G. (2004). The chemical recycling of PET in the framework of sustainable development, Water, Air, & Soil Pollution: Focus, Vol 4, No. 4-5, (October 2004), pp. 385-396, ISSN 1567-7230 Aguado, J. & Serrano D. (1999). Feedstock Recycling of Plastic Wastes, The Royal Society of Chemisty, ISBN 0-85404-531-7, United Kingdom
Al-Salem, S. (2009). Establishing an integrated databank for plastic manufacturers and converters in Kuwait, Waste Management, Vol. 29, No. 1, (January 2009), pp. 479-484 Al-Salem, S., Lettieri, J., Baeyens, J. (2009) Recycling and recovery routes of plastic solid waste (PSW): A review, Waste Management, Vol. 29, No. 10, (October 2009), pp. 2625-2643 Alter, H. (1986). Disposal and Reuse of Plastics, In: Encyclopedia of Polymer Science and Engineering, pp. 103-128, Herman Mark, Wiley Interscience, ISBN 978-0471880981 New York



